General input parameters¶
- Notes about format
- Dispersion relations
dispersion_interpolatedispersion_interpolate_samplingdispersion_interpolate_step_sizedispersion_interpolate_methoddispersion_interpolate_typedispersion_velocities_numdiffdispersion_write_preinterdispersion_write_postinterdispersion_write_startdispersion_write_enddispersion_num_kpoints_along_linedispersion_effmassdispersion_effmass_diagonalizedispersion_effmass_transform
- Electron transport
transport_calctransport_methodtransport_integration_methodtransport_integration_spectral_smearingtransport_integration_spectral_densitytransport_integration_spectral_energy_cutofftransport_chempot_mintransport_chempot_maxtransport_chempot_samplestransport_energycutbandtransport_include_bandstransport_use_analytic_scatteringtransport_drop_valencetransport_drop_conduction
- Density of states
- General parameters
temperature_mintemperature_maxtemperature_stepsgamma_centermaxeintocc_cutoffe_fermi_in_gape_fermie_vbme_shiftskw_expansion_factorcarrier_valence_energycarrier_conduction_energycarrier_dos_analytickdefect_ionizationdonor_numberdonor_degen_factdonor_energyacceptor_numberacceptor_degen_factacceptor_energyreadreadfilescissorsympreclibinfoonlytotalrateparallelrun_tests
Notes about format¶
The input files follow normal YAML conventions.
Please inspect the sample file input/param.yml.
Even though many parameters have default values if not
specified the user should always run the calculations with
fully specified input files for consistency and reproducibility.
Dispersion relations¶
The following parameters are related to the energy and velocity dispersion relations.
dispersion_interpolate¶
If set to True the band structure is interpolated on a
k-point grid.
Example:
dispersion_interpolate: False
Do not interpolated the band structure.
dispersion_interpolate_sampling¶
The target k-point sampling when performing interpolation.
Example:
dispersion_interpolate_sampling: [45,45,45]
Interpolates the input band structure to a grid density of 45, 45 and 45 k-points along the unit axis of the supplied k-point grid.
dispersion_interpolate_step_size¶
The target k-point step size in inverse AA. In order for this parameter to work, the user have to set
dispersion_interpolate_sampling: [0,0,0]
Example:
dispersion_interpolate_sampling: [0.1,0.1,0.1]
Creates a k-point sampling that is at least as dense as to give a step size of 0.1 inverse AA between each k-point along each reciprocal axis.
dispersion_interpolate_method¶
Choses which interpolative method to use. The following options are currently available:
- linearnd - Uses
LinearNDInterpolatorin SciPy. - interpn - Uses
interpnin Scipy. - rbf - Uses the Scipy version, but that is memory intensive
- wildmagic - Uses the GeometricTools (former WildMagic) interpolation routines.
- skw - Uses Fourier interpolation
- tb - Extracts the energies on a denser grid from a tight- binding model
Tests have shown that the last three methods are quite general and, given what they are, quite accurate.
Example:
dispersion_interpolate_method: "wildmagic"
Will for instance use the WildMagic library.
dispersion_interpolate_type¶
Additional selective layer for the method chosen by :ref’dispersion_interpolate_method. Currently, the following options are availble:
- nearest or linear - if
dispersion_interpolate_method= linearnd - trilinear, tricubic_exact, tricubic_bspline, akima
- if
dispersion_interpolate_method= wildmagic
Example:
dispersion_interpolate_type: "akima"
Uses the Akima interpolation in the WildMagic library.
dispersion_velocities_numdiff¶
Use numerical differentiation to calculate the velocities if they are not present on entry, or/and use numerical differentiation to extract the velocities after the dispersions have been interpolated (used by default for the interpolat routines that do not support velocity extraction)
Example:
dispersion_velocities_numdiff: False
Turns for instance of the numerical difference calculation of the velocities. In this case please make sure that the velocities are present on input or that they are genrated by other means.
dispersion_write_preinter¶
Selects if a line extraction of the band structure is written to
the file bands before interpolation. If velocities are present
this is also written to the file velocities
Example:
dispersion_write_preinter: False
Writes the extracted band structure values along a line to file(s).
dispersion_write_postinter¶
Selects if a line extraction of the band structure is written to
the file bands_inter after interpolation. If velocities
are present this is also written to the file velocities_inter
Example:
dispersion_write_postinter: False
Does not write the extracted band structure values along a line to file(s).
dispersion_write_start¶
The start point (in direct coordinates) for the line extraction.
Example:
dispersion_write_start: [0.0, 0.0, 0.0]
An example start point, here the Gamma point.
dispersion_write_end¶
The end point (in direct coordinates) for the line extraction.
Example:
dispersion_write_end: [0.5, 0.0, 0.0]
dispersion_num_kpoints_along_line¶
How many samples to use along the line to be extracted.
Example:
dispersion_num_kpoints_along_line: 20
Here 20 points is used along the line.
dispersion_effmass¶
Calculate the effective mass tensor along the unit vectors of the configured reciprocal cell. The resulting tensor is in units of the free electron mass. Currently it is not printed out and an error will occur.
Example:
dispersion_effmass: False
Do not calculate the effective mass tensor.
dispersion_effmass_diagonalize¶
Diagonalize the calculated effective mass tensor. Currently the diagonal elements and the eigenvectors are not printed out and an error will occur.
Example:
dispersion_effmass_diagonalize: False
Do not diagonalize the effective mass tensor.
dispersion_effmass_transform¶
The transformation vectors for the effective mass tensor. The elements [0,:] give the first vector, [1,:] the second and [2,:] the third. Should be given in direct coordinates. If the array is left empty, no transformation is performed.
Example:
dispersion_effmass_transform: []
Do not transform the effective mass tensor.
Electron transport¶
The following parameters determines how the transport of electrons is to be determined.
transport_calc¶
Determines if the transport calculations are to executed.
Example:
transport_calc: True
Calculate the transport properties.
transport_method¶
Selects which mode to use to calculate the transport properties. Currently three different modes are accepted;
- closed - The integrals are solved using the closed Fermi-Dirac integrals. Only available if the band structure is generated by means of analytic models. Only one scattering mechnism can be used for each band in this approach.
- numeric - A numerical integration of the Fermi-Dirac integrals, which allows to concatenate different scattering mechanisms for each band.
- numerick - The integrals are solved by integrating over the k-point grid or by utilizing the spectral function.
Example:
transport_method: "numerick"
In this example the transport integrals are solved using the closed analytical expressions for the Fermi-Dirac integrals.
transport_integration_method¶
Selects which method to use for solving the integral over the k-points.
Only applicable if transport_method is set to numerick.
- trapz - Use the trapezoidal integration scheme implemented in SciPy
- simps - Use the Simpson integration scheme implemented in SciPy
- romberg - Use the Romberg integration scheme implemented in SciPy
- tetra - Use the linear tetrahedron method
- smeared - Use the weighted sum approach with a smearing factor
transport_integration_spectral_smearing¶
Gaussian smearing factor for the weighted sum approach.
In units of eV. Only relevant if transport_integration_method
is set to smeared.
Example:
transport_integration_spectral_smearing: 0.1
Would set it to 0.1 eV.
transport_integration_spectral_density¶
The sampling density of the spectral function. Only relevant if
transport_integration_method is set to tetra or smeared.
Example:
transport_integration_spectral_density: 1000
An example requesting 1000 samples.
transport_integration_spectral_energy_cutoff¶
Determines the extra padding that is used for the spectral function on
both sides of the requested chemical potential. If multiple chemical
potentials are requested, the lowest and the highest value is checked and
the range of the energy interval on which the spectral function is
calculated is padded with the specified value. Only relevant if
transport_integration_method is set to tetra or smeared. In
units of eV.
Example:
transport_integration_spectral_energy_cutoff: 1.0
Here, 1.0 eV is subtracted (added) to the smallest (largest) requested chemical potential.
transport_chempot_min¶
The minimum chemical potential requested for which the transport coefficients are calculated. In units of eV.
Example:
transport_chempot_min: -1.0
Starts the calculation of the transport properties at -1.0 eV.
transport_chempot_max¶
The maximum chemical potential requested for which the transport coefficients are calculated. In units of eV.
Example:
transport_chempot_max: 1.0
Ends the calculation of the transport properties at 1.0 eV.
transport_chempot_samples¶
The number of chemical potential samples to use between
transport_chempot_min and transport_chempot_max.
Example:
transport_chempot_samples: 100
Extract the transport coefficients at 100 points between
transport_chempot_min and transport_chempot_max.
transport_energycutband¶
Bands that reside transport_energycutband outside
the chemical potential is dropped from the calculation of the
transport coefficients. All k-points
are currently analyzed in order to determine which bands fall inside
the energy range
[transport_chempot_min-transport_energycutband,``transport_chempot_max``+``transport_energycutband``]
. Units in eV.
Example:
transport_energycutband: 1.0
Substract and add 1.0 eV to transport_chempot_min and
transport_chempot_max, respectively. Bands that does not have
any k-point with energy in the range [-2.0 eV, 2.0 eV] is not included
in the calculation of the transport coefficients.
transport_include_bands¶
A list containing specific bands on which to calculate the transport
coefficients. If the list is empty, use all bands within the range set by
:ref:transport_energycutband. Band index starts at 1.
Example:
transport_include_bands: [3, 4, 10]
Calculate the transport coefficients for band 3, 4 and 10.
transport_use_analytic_scattering¶
Determines if the analytic parabolic scattering models should be used. They can be applied also to dispersions which are not parabolic, but such an application have to be physically justified.
Example:
transport_use_analytic_scattering: False
Use the density-of-states to set up the scattering mechanisms.
transport_drop_valence¶
Determines if all valence band should be dropped while reading e.g. external data. Currently only works for the VASP interface.
Example:
transport_drop_valence: False
Do not exclude the valence bands during read-in.
transport_drop_conduction¶
Determines if all conduction bands should be dropped while reading e.g. external data. Currently only works for the VASP interface.
Example:
transport_drop_conduction: False
Do not exclude the conduction bands during read-in.
Density of states¶
Here follows input parameters related to the calculation of the density of states.
dos_calc¶
Determines if the user wants to calculate the density of states.
Even if this flag is set to False, the density of states is
sometimes calculated if needed, e.g. if the density of states
dependent scattering models are employed. However, with this
parameter set to True and e.g. transport_calc set to
False it is possible to only calculate the density of states.
dos_calc: False
Do not calculate the density of states.
dos_e_min¶
The minimum energy to use for the density of states calculation. In units of eV. The reference is with respect to the aligned Fermi level and consequetive shift that might have been applied. Note that the range of density of states calculation might change if it is called from other routines, e.g. the density of states dependent scattering models in order to cover enough energies.
dos_e_min: -5.0
Calculate the density of states from -5.0 eV.
dos_e_max¶
The maximum energy to use for the density of states calculation. In units of eV. The reference is with respect to the aligned Fermi level and consequetive shift that might have been applied. Note that the range of density of states calculation might change if it is called from other routines, e.g. the density of states dependent scattering models in order to cover enough energies.
dos_e_max: 2.0
Calculate the density of states to 2.0 eV.
dos_num_samples¶
The number of energy samples between dos_e_min and
dos_e_max.
dos_num_samples: 1000
Use 1000 energy points from dos_e_min to dos_e_max.
dos_smearing¶
Gaussian smearing factor in units of eV. Only relevant if
dos_integrating_method is set to smeared, trapz,
simps or romb.
dos_smearing: 0.1
dos_integrating_method¶
Determines which method of integration to use to obtain the density of states. The following options are available:
- trapz - trapezoidal integration
- simps - Simpson integration
- romb - Romberg integration
- tetra - linear tetrahedron method without Blochl corrections
dos_integrating_method: "trapz"
Use trapezoidal integration to obtain the density of states.
General parameters¶
Here follows general parameters.
temperature_min¶
The minimum temperature in K.
Example:
temperature_min: 100
The minimum temperature is set at 100 K.
temperature_max¶
The maximum temperature in K.
Example:
temperature_max: 700
The maximum temperature is set at 700 K.
temperature_steps¶
The number of temperature steps from temperature_min
to temperature_max.
Example:
temperature_steps: 7
In total 7 temperature steps, resulting in temperature samplings at 100, 200, 300, 400, 500, 600 and 700 K.
gamma_center¶
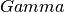 centered k-point grids? Anything else is currently
not supported (or tested).
centered k-point grids? Anything else is currently
not supported (or tested).
Example:
gamma_center: True
Notifies that the k-point grids are centered.
maxeint¶
The limites of the dimensionless carrier energy 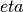 used for the numerical solution of the Fermi-Dirac integrals.
Only relevant if
used for the numerical solution of the Fermi-Dirac integrals.
Only relevant if transport_method is set to numerick.
Example:
maxeint: 100
Sets the limits of the Fermi-Dirac integrals to 100 .
occ_cutoff¶
The cutoff to use when detecting occupancies. Used for detecting the valence band maximum, conduction band minimum and then also for the band gap.
Example:
occ_cutoff: 1.0e-4
The occupancy cutoff is set at 1.0e-4, which means that states with an occupancy less than this will be assumed not occupied and vice versa.
e_fermi_in_gap¶
Determines if the Fermi level is to be placed in the middle of the gap.
Example:
e_fermi_in_gap: False
Do not place the Fermi level in the middle of the gap.
e_fermi¶
Determine if one should shift the energies to the supplied Fermi level (usually read in the interface).
Example:
e_fermi: True
Shift the energies such that zero is placed at the supplied Fermi level.
e_vbm¶
Determines if to set the Fermi level at the valence band maximum.
Example:
e_vbm: False
Do not set the Fermi level at the top valence band.
e_shift¶
After all alignments have been performed, perform this additional shift. Units in eV.
Example:
e_shift: 0.0
Sets the additional energy shift to 0 eV.
skw_expansion_factor¶
The expansion factor used in the SKW routine. It is basically
tells how many unit cells that can be used. Only relevant if
dispersion_interpolate_method is set to skw.
Example:
skw_expansion_factor: 5
Use 5 unit cells in each direction. In a second step a sphere is cut from this volume, thus removing the points in the far corners of this volume in the interpolation procedure.
carrier_valence_energy¶
The cutoff in which where to interpret the carriers as p-type. Used in the calculation of the carrier concentration. Units in eV.
Example:
carrier_valence_energy: 0.0
Would make sure all carriers at negative energies are interpreted as p-type.
carrier_conduction_energy¶
The cutoff in which where to interpret the carriers as n-type. Used in the calculation of the carrier concentration. Units in eV.
Example:
carrier_valence_energy: 0.0
Would make sure all carriers at positive energies are interpreted as n-type.
carrier_dos_analytick¶
Determines if the carrier concentration should be recaculated after being set up with analytical models. Only relevant if the band structure is generated from analytical models.
Example:
carrier_dos_analytick: True
Do not recalculate and use the analytical expressions for the carrier concentration.
defect_ionization¶
Determines if we shoudl use the expressions for the defect ionization in order to calculate the p- and n-type carrier concentration.
Example:
defect_ionization: False
Do not use the models for the defect_ionization to adjust the p- and n-type carrier concentration.
donor_degen_fact¶
The degeneracy factor for the donors.
Example:
donor_degen_fact: 0.75
A degeneracy factor of 0.75 is used.
donor_energy¶
The energy of the donor in units of eV. Should be referenced to the energy after all adjustments to the Fermi level and additional energy shifts have been performed.
Example:
donor_energy: 0.0
The donor energy is 0 eV.
acceptor_number¶
The density of acceptors in units of 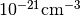 .
.
Example:
donor_number: 0.0
No acceptors present.
acceptor_degen_fact¶
The degeneracy factor for the acceptors.
Example:
acceptor_degen_fact: 0.75
A degeneracy factor of 0.75 is used.
acceptor_energy¶
The energy of the acceptor in units of eV. Should be referenced to the energy after all adjustments to the Fermi level and additional energy shifts have been performed.
Example:
acceptor_energy: 0.0
The acceptor energy is 0 eV.
read¶
Determine how to set up the band structure and/or how to read data. The following options are possible:
param - The band structure is generated from the parameter files. For all cases the band structure is generated by analytical models. The parameters pertaining to the construction of the bandstructure itself is set in the file
bandparam.yml.numpy - Read data from NumPy datafiles without group velocities.
The datastructure of the supplied numpy arrayshould be on the following format:[[kx], [ky], [kz], [e_1], [v_x_1], [v_y_1], [v_z_1],[e_2], [v_x_2], [v_y_2], [v_z_2], … ,[e_n], [v_x_n], [v_y_n], [v_z_n]]The band parameters still need to be set in
bandparam.ymlas they contain necessary information about scattering etc.numpyv - Read data from NumPy datafiles, including group velocities.
The datastructure of the supplied numpy arrayshould be on the following format:[[kx], [ky], [kz], [e_1], [e_2], … , [e_n]]The band parameters still need to be set in
bandparam.ymlas they contain necessary information about scattering etc.vasp - Read data from a supplied VASP XML file, typically vasprun.xml. The band parameters still need to be set in
bandparam.ymlas they contain necessary information about scattering etc.
Example:
read: param
Construct the band structure from the parameters present in
bandparam.yml.
readfile¶
The name of the file to be read. Depending on read it has the
following behaviour:
- param - not relevant
- vasp - the name of the VASP XML file, if not set it defaults to vasprun.xml
- numpy - the name of the NumPy datafile
- numpyv - the name of the NumPy datafile
Example:
readfile: ""
Use defaults, e.g. vasprun.xml for VASP.
scissor¶
Apply a simple scissor operator to increase the band gap. Only works of the band gap has been correctly determined. In units of eV if not False.
Example:
scissor: False
Do not apply a scissor operator.
symprec¶
The symmetry cutoff parameters. Passed to Spglib. VASP also uses an internal symmetry parameter which is called SYMPREC. Spglib need to reproduce the symmetry that was detected in VASP in order for the k-point grids and thus the mapping between the IBZ and BZ to be valid. If errors regarding this is invoked, please try to adjust symprec.
Example:
symprec: 1.0e-6
If two coordinates are within 1.0e-6 it is assumed that they are the same and symmetry is thus detected.
libinfo¶
Determines if printout to stdout is performed in the interfaces to the external libraries.
Example:
libinfo: False
Do not print stdout information from the interfaces.
onlytotalrate¶
Determines if the users wants to store the relaxation time for each scattering mechanism. This is usefull for visualization purposes, but is simply very memory demanding. Users should try to leave this to True.
Example:
onlytotalrate: True
Only store the total relaxation time.